Searching for COVID-19 treatments using metabolic networks
Apostolos Chalkis introduces dingo, a package for the analysis of metabolic networks, on which he worked as part of this Tweag fellowship. This article republishes Apostolos’ blog post from Tweag’s website. The original blog post can be found here.
As part of my Tweag fellowship, I developed dingo,
a Python package to analyze metabolic networks. dingo provides three methods
that complement the openCOBRA project. The main
advantage of dingo is the efficient implementation
of new sampling algorithms that generate steady states of metabolic networks.
In this post, I’ll explain what this all means and even discuss how
we can use this tool to discover new COVID-19 treatments.
Metabolic networks and COVID-19
Every moment, in every cell of our body, thousands of chemical reactions are taking place. In cells infected with a virus, some of these reactions benefit the virus by producing molecules—metabolites—that are critical for making new virus particles. Many of these reactions are also critical for our own survival, but if we can find one or two reactions that are more important for the virus than for ourselves, inhibiting these reactions may become an effective antiviral treatment.
Alina Renz, Lina Widerspick, and Andreas Dräger did exactly this for SARS-CoV-2 in their recent paper. They identified the following reaction as a promising target:
\[(\textbf{GK1}):\ ATP + GMP \rightarrow ADP + GDP\]Don’t worry if your high-school chemistry is rusty: the above notation simply says that GK1 (named after the enzyme guanylate kinase) is a reaction that turns a pair of molecules called ATP and GMP into another pair of molecules, ADP and GDP. What these molecules are, exactly, won’t concern us here (though you may get a kick out of the fact that these molecules are directly related to the ‘A’ and ‘G’ letters of your DNA). Instead, our goal is to
- understand the method by which Renz et al. identified this particular reaction out of thousands of others, and
- see how we can do the
same (and perhaps a bit more?) using
dingo, the new Python package I mentioned in the beginning.
Apart from GK1, the reaction shown above, ADP and other metabolites take part in many other reactions. Together all these metabolites and reactions form a metabolic network.

A small fragment of the human metabolic network (Recon1)
Generally, the concentrations of metabolites change with time. For instance, GK1 increases the concentration of ADP, whereas some other reactions—those for which ADP is an input rather than an output—decrease it. It may also happen that all reactions that affect ADP exactly balance each other out so that the concentration of ADP doesn’t change over time. If this happens for every single metabolite, not just ADP, then we say that the metabolic network is in a steady state.
There is an infinite number of steady states. Each steady state specifies a particular flux for every reaction—that is, the rate at which the reaction is carried out. A common way to study a metabolic network is to look for the steady state that maximizes some objective function—for instance, the rate at which new virus particles are produced. This is called the Flux Balance Analysis, or FBA, and is the primary method used by Renz et al. to analyze their human-virus model.
Renz et al. compute two optimal steady states using FBA: one that maximizes the virus’s growth rate, and another that maximizes the human human’s biomass maintenance. Then they identify as potential anti-viral targets the reactions whose fluxes differ significantly between these two steady states.
How dingo can help
The dingo package is a part of the
GeomScale organization, and the sampling methods it uses are developed in GeomScale’s package volesti. The main method that dingo uses is called Multiphase Monte Carlo Sampling (MMCS) algorithm, and you can read about it in this paper. However, it also provides FBA method.
The following python code uses dingo to compute these two optimal steady states with FBA. To load the human-virus model with dingo, I loaded the model with open COBRA toolbox and used the dingo’s matlab script to extract the .mat file that dingo can load.
from dingo import MetabolicNetwork
# load the model
model = MetabolicNetwork.from_mat('path/to/iAB_AMO1410_SARS_CoV_2.mat')
# get information about the biomass functions
covid_biomass_index = model.biomass_index
human_biomass_index = covid_biomass_index - 1
# perform FBA when the objective function represents the virus' growth rate
fba_for_covid_optimized_biomass = model.fba()
# set the objective function to be the human's biomass function
biomass_function = np.zeros(model.num_of_reactions)
biomass_function[human_biomass_index] = 1
model.set_biomass_function(biomass_function)
# perform FBA when the objective function is the human's biomass function
fba_for_human_optimized_biomass = model.fba()
In the figure below, I plot the fluxes of four reactions for the human’s biomass- and virus-optimal steady states. The reaction GK1 is part of the purine metabolism. The reaction CTPS2 produces certain amino acids, which are the building blocks of proteins. The reaction 3DSPHR participates in sphingolipid metabolism, while the reaction TYMSULT is related to the protein-tyrosine sulfotransferase activity.
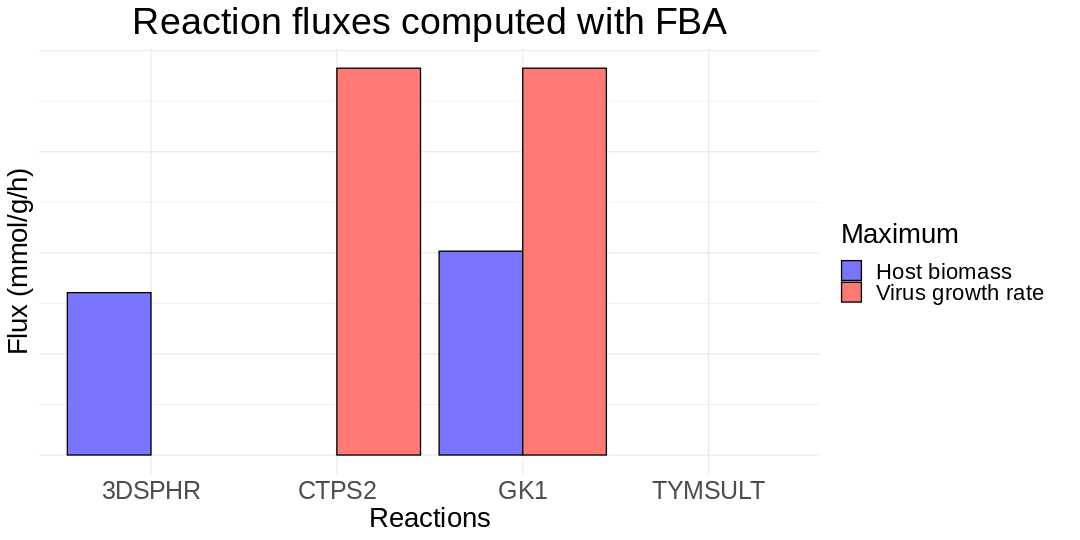
For both GK1 and CTPS2, FBA implies that the fluxes increase when the virus biomass takes its maximum possible value. That happens because these reactions are related to the growth of an organism. Thus, when the human is infected by SARS-CoV-2, the activity of those reactions increases to meet the new needs of the organism. Moreover, when Renz et al. experimentally turned off the GK1 reaction, the biomass of the virus decreased drastically without affecting human’s organism. This is why they recognize it as potential anti-viral target.
For 3DSPHR, FBA implies that its flux will fall to zero when the virus’ biomass takes its maximum value. On the other hand, for TYMSULT, FBA implies that the flux will be equal to zero for both optimized cases.
However, even after imposing an objective function, there still remain an infinite number of optimal steady states, and FBA computes only a single one of them. Even if the biological system is in a state close to an optimal one, it may select a different optimal state from the one found by FBA.
Flux sampling with dingo
dingo can perform an alternative, Bayesian-like analysis of steady states. For this, we
assume that each optimal steady state has the same probability to be chosen by the
biological system. Then, dingo generates a large number of optimal steady states using a
Markov Chain Monte Carlo sampling algorithm. Instead of a single flux value, we can
now compute credible intervals, estimate the average value, or employ other statistical
methods to express our uncertainty about the flux.
Here’s how flux sampling could provide more information than FBA
about the fluxes. I am going again to use the same 4 reactions as before. The
following python code uses dingo to generate the data to estimate the probability distribution
of any reaction flux of the model, for both human’s biomass- and virus-optimized cases.
from dingo import MetabolicNetwork, PolytopeSampler
# load the model
model = MetabolicNetwork.fom_mat('path/to/iAB_AMO1410_SARS_CoV_2.mat')
# get information about the biomass functions
covid_biomass_index = model.biomass_index
human_biomass_index = covid_biomass_index - 1
# sample optimal steady states when the objective function represents the virus' growth rate
sampler = polytope_sampler(model)
steady_states_covid_max_biomass = sampler.generate_steady_states()
# set the objective function to be the human's biomass function
biomass_function = np.zeros(model.num_of_reactions)
biomass_function[human_biomass_index] = 1
model.set_biomass_function(biomass_function)
# sample optimal steady states when the objective function is the human's biomass function
sampler = PolytopeSampler(model)
steady_states_human_max_biomass = sampler.generate_steady_states()
Let’s take a look at the estimated probability densities for the 4 reaction fluxes. I also mark with
two vertical dotted lines the two fluxes that I previously computed with FBA. Next, we compare FBA’s fluxes
with the average fluxes we get from dingo’s sampling routines.

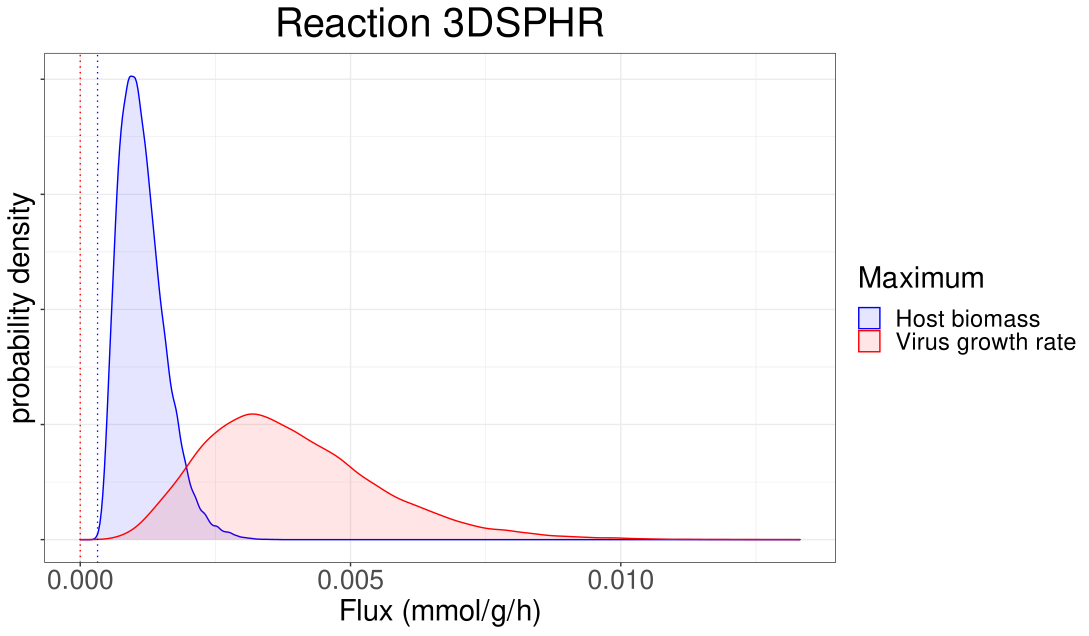


The blue density is computed by human’s biomass-optimized states, while the red density is computed by virus-optimized states. The fluxes with a higher value on the y-axis in the plot are more possible to be selected by the metabolic network.
For all the reactions the probability densities reveal that the fluxes computed by FBA are very unlikely to be selected by the metabolic network. In particular, FBA computes really extreme values for all the fluxes; the probability density values of those fluxes are very small or even close to zero. Moreover, let’s make some interesting comments on these four plots.
For the reaction TYMSULT, both FBA and flux sampling imply that
the fluxes would have small value. However, dingo samples fluxes whose values differ
from zero and also provides more statistical information implied by the
estimated probability densities.
The 3DSPHR flux densities imply that with high probability the flux in human’s biomass-optimized case is smaller than the flux in the virus-optimized case. Moreover, the average flux in the human’s biomass-optimized case is smaller than that in the virus-optimized case, while FBA implies the opposite.
For both CTPS2 and GK1, flux sampling again reveals that the fluxes computed via FBA are very unlikely to be selected. However, it agrees with FBA about the fluxes’ order of magnitude; the average flux in the virus-optimized case equals two times the flux in the human’s biomass-optimized case.
To further illustrate those differences, in the following figure, I plot the average fluxes we
get from the estimated probability densities computed with flux sampling. Notice that
this bar plot is different from the previous one, which dingo computed with FBA. The flux
values of TYMSULT are not zero here, as we noticed in the corresponding probability
densities, but they are both much smaller than the other flux values.

To summarize, flux sampling could agree or disagree with FBA about the flux values. In any case, the flux densities we estimate provide us with more statistical information and with more accurate expected flux values than FBA. However, the analysis of those results is not always an easy task. Furthermore, we don’t have any guarantee that flux-sampling will lead to a more sophisticated result comparing to FBA. But at least we have more information to analyze.
Two more advantages of dingo
Unlike FBA, dingo doesn’t require the assumption that the human’s or virus’s
biomass production rate takes its maximum value. This way, we can study the
complete set of steady states. In addition, I will show how we can use the
generated steady states to correlate the fluxes of two reactions or the value of
biomass with any flux. I’ll illustrate both advantages with a single example.
The following python code generates steady states without imposing any objective function on them.
from dingo import MetabolicNetwork, PolytopeSampler
# load the model
model = MetabolicNetwork.fom_mat('path/to/iAB_AMO1410_SARS_CoV_2.mat')
# set the unbiased objective function
biomass_function = np.zeros(model.num_of_reactions)
model.set_biomass_function(biomass_function)
# generate steady states
sampler = polytope_sampler(model)
steady_states = sampler.generate_steady_states()
To capture the dependency between the biomass production and a flux, we use a 2D dimensional copula. A 2D copula is a bivariate distribution whose each marginal distribution is uniform. For more details about what a copula is, you can read this paper. Thus, I plot a copula to capture the dependency between the human’s biomass production rate and the flux of the reaction GLGNS1, which converts glucose into glycogen and it is crucial for the growth rate of an organism.
We notice that the most steady states correspond to either slow human biomass production and small GLGNS1 flux or fast biomass production and large GLGNS1 flux. This illustrates the crucial role that GLGNS1 plays for organism growth.
Conclusion
In this blog post, I showed how we can use the python package dingo to analyze metabolic networks. Focusing on the model of Renz, Widerspick, and Dräger, I illustrated how dingo can provide additional statistical information about a metabolic network compared to other standard methods such as FBA. I hope that the efficiency of dingo could be an important step to overcome the computational barriers that have existed until today to study metabolic networks using high dimensional MCMC sampling.
Acknowledgements
Many thanks to my Tweag mentors Roman Cheplyaka and Simeon Carstens for their helpful and crucial comments and contributions to this blog post and to my coding project during my Tweag fellowship. I also would like to thank Alina Renz, Lina Widerspick, and Andreas Dräger for their useful comments on this blog post. Last but not least, I would like to thank my collaborators from GeomScale org, namely Vissarion Fisikopoulos, Elias Tsigaridas, and Haris Zafeiropoulos for their useful comments and contributions throughout my Tweag fellowship.